Case
V.P. is a 13-year-old female with no past medical history who presented to an outside hospital with a 3-week history of fatigue, emesis, 10-pound weight loss, headache, cough, and intermittent shortness of breath. She denied fevers, respiratory symptoms, or diarrhea. Initial lab work-up showed a normal basic metabolic panel, slightly elevated total bilirubin, and mildly elevated ALT and AST. Abdominal ultrasound was negative for gallstones. She was admitted to the GI service, but had worsening respiratory distress with chest tightness and desaturations. ECG showed low voltages and a wide QRS complex. She was transferred to the CICU at CHOP.
On exam, she was tachypneic, with difficulty lying flat. Breath sounds were decreased at the bases. Patient was tachycardic with normal S1, S2, an audible S3 gallop, and no murmurs. Extremities were warm and well perfused. Abdomen was mildly tender and obese, making it difficult to assess for hepatomegaly. Echocardiogram showed severe LV dysfunction, SF 10%. Inotropic medications and diuretics were initiated. Viral blood studies were negative. The patient required CPAP for worsening respiratory distress. Due to worsening heart failure symptoms refractory to medical management, a ventricular assist device (VAD) was implanted. During implant, a core of the LV was sent for tissue PCR and pathology. V.P. stabilized on the VAD, though she was having PACs, PVCs, and intermittent atrial rhythms.
Discussion
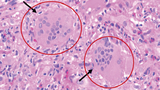
Pathology revealed giant cell myocarditis (GCM), evidenced by significant myocyte loss and fibrosis, and chronic inflammatory infiltrate composed of lymphocytes, eosinophils, and plasma cells with focal giant cells. (See Figure 1.)
Giant cell myocarditis is a rare but often fatal form of myocarditis. The clinical course is usually a rapid deterioration in cardiac function over several days to weeks. Ventricular tachycardia and heart block occur in a large number of GCM patients. Unlike fulminant lymphocytic myocarditis, which has a very good prognosis, GCM has a poor prognosis with many patients either dying or undergoing heart transplantation in the first year after diagnosis. Unfortunately, recurrence of giant cells in the transplanted heart is estimated to be 20% to 25%.
GCM is attributed to T lymphocyte-mediated inflammation of the heart muscle and is associated with systemic autoimmune diseases in approximately 20% of cases. Medical treatment is aimed at attenuating T-cell function and includes combination immune suppression with cyclosporine, prednisone, and azathioprine. These medications have been associated with prolonged transplant-free survival in patients with acute GCM. There are currently no good data to guide immune suppression for long-term maintenance of remission, though continued treatment appears important, as cessation of immune suppression may lead to fatal relapse.
Our patient, V.P., was started on cyclosporine, azathioprine, and prednisone once diagnosed with GCM. There was no significant improvement in heart function, though she remained well supported on the VAD. She was listed for transplant and discharged on VAD 5 weeks after presentation. She returned to clinic regularly.
Five months after diagnosis, V.P. underwent successful heart transplantation at CHOP. She received 3 days of high-dose IV steroids and 3 doses of antithymocyte globulin (ATG). Her immune suppression medications were switched to tacrolimus and mycophenolate. A heart biopsy 2 weeks after transplant showed no rejection and no giant cells in the new heart. Less than 3 weeks after transplant, V.P. went home. A routine biopsy done 1 month post-transplant showed no evidence of rejection, but did show recurrence of giant cells in the transplanted heart, and V.P. was readmitted for more high-dose steroids and ATG.
Routine heart biopsies were conducted at 8 and 12 weeks post-transplant, which showed no evidence of rejection and no presence of giant cells. At 6 months post-transplant, VP complained of “palpitations.” Her troponin-I was elevated at 0.64, and heart biopsy revealed presence of giant cells, but no rejection. She was admitted and treated with IV steroids (1g/day) for 3 days and started on an additional immune suppressant, sirolimus.
Approximately 9 months after transplant, V.P. underwent placement of an implantable cardioverter defibrillator (ICD), as she had increasing runs of tachyarrhythmias, commonly seen with GCM.
V.P. has undergone heart biopsies at 9 months, 12 months, and 18 months post-transplant, all of which have been negative for rejection and giant cells.
Currently, V.P. returns for routine transplant visits every 3 months. She has normal heart function and takes medications twice daily. She will undergo annual heart biopsies, or more frequently if there are changes on echocardiogram, increase in troponin or BNP, or symptoms. V.P. does very well in school and participates in many activities both in and outside of school.
Featured in this article
Specialties & Programs
Case
V.P. is a 13-year-old female with no past medical history who presented to an outside hospital with a 3-week history of fatigue, emesis, 10-pound weight loss, headache, cough, and intermittent shortness of breath. She denied fevers, respiratory symptoms, or diarrhea. Initial lab work-up showed a normal basic metabolic panel, slightly elevated total bilirubin, and mildly elevated ALT and AST. Abdominal ultrasound was negative for gallstones. She was admitted to the GI service, but had worsening respiratory distress with chest tightness and desaturations. ECG showed low voltages and a wide QRS complex. She was transferred to the CICU at CHOP.
On exam, she was tachypneic, with difficulty lying flat. Breath sounds were decreased at the bases. Patient was tachycardic with normal S1, S2, an audible S3 gallop, and no murmurs. Extremities were warm and well perfused. Abdomen was mildly tender and obese, making it difficult to assess for hepatomegaly. Echocardiogram showed severe LV dysfunction, SF 10%. Inotropic medications and diuretics were initiated. Viral blood studies were negative. The patient required CPAP for worsening respiratory distress. Due to worsening heart failure symptoms refractory to medical management, a ventricular assist device (VAD) was implanted. During implant, a core of the LV was sent for tissue PCR and pathology. V.P. stabilized on the VAD, though she was having PACs, PVCs, and intermittent atrial rhythms.
Discussion
Pathology revealed giant cell myocarditis (GCM), evidenced by significant myocyte loss and fibrosis, and chronic inflammatory infiltrate composed of lymphocytes, eosinophils, and plasma cells with focal giant cells. (See Figure 1.)
Giant cell myocarditis is a rare but often fatal form of myocarditis. The clinical course is usually a rapid deterioration in cardiac function over several days to weeks. Ventricular tachycardia and heart block occur in a large number of GCM patients. Unlike fulminant lymphocytic myocarditis, which has a very good prognosis, GCM has a poor prognosis with many patients either dying or undergoing heart transplantation in the first year after diagnosis. Unfortunately, recurrence of giant cells in the transplanted heart is estimated to be 20% to 25%.
GCM is attributed to T lymphocyte-mediated inflammation of the heart muscle and is associated with systemic autoimmune diseases in approximately 20% of cases. Medical treatment is aimed at attenuating T-cell function and includes combination immune suppression with cyclosporine, prednisone, and azathioprine. These medications have been associated with prolonged transplant-free survival in patients with acute GCM. There are currently no good data to guide immune suppression for long-term maintenance of remission, though continued treatment appears important, as cessation of immune suppression may lead to fatal relapse.
Our patient, V.P., was started on cyclosporine, azathioprine, and prednisone once diagnosed with GCM. There was no significant improvement in heart function, though she remained well supported on the VAD. She was listed for transplant and discharged on VAD 5 weeks after presentation. She returned to clinic regularly.
Five months after diagnosis, V.P. underwent successful heart transplantation at CHOP. She received 3 days of high-dose IV steroids and 3 doses of antithymocyte globulin (ATG). Her immune suppression medications were switched to tacrolimus and mycophenolate. A heart biopsy 2 weeks after transplant showed no rejection and no giant cells in the new heart. Less than 3 weeks after transplant, V.P. went home. A routine biopsy done 1 month post-transplant showed no evidence of rejection, but did show recurrence of giant cells in the transplanted heart, and V.P. was readmitted for more high-dose steroids and ATG.
Routine heart biopsies were conducted at 8 and 12 weeks post-transplant, which showed no evidence of rejection and no presence of giant cells. At 6 months post-transplant, VP complained of “palpitations.” Her troponin-I was elevated at 0.64, and heart biopsy revealed presence of giant cells, but no rejection. She was admitted and treated with IV steroids (1g/day) for 3 days and started on an additional immune suppressant, sirolimus.
Approximately 9 months after transplant, V.P. underwent placement of an implantable cardioverter defibrillator (ICD), as she had increasing runs of tachyarrhythmias, commonly seen with GCM.
V.P. has undergone heart biopsies at 9 months, 12 months, and 18 months post-transplant, all of which have been negative for rejection and giant cells.
Currently, V.P. returns for routine transplant visits every 3 months. She has normal heart function and takes medications twice daily. She will undergo annual heart biopsies, or more frequently if there are changes on echocardiogram, increase in troponin or BNP, or symptoms. V.P. does very well in school and participates in many activities both in and outside of school.
Contact us
Pediatric Transplant Center