Gender Discrepancy Case Study
Maternal history
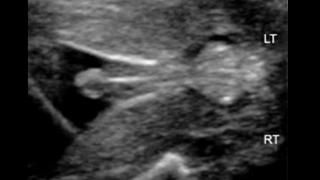 Ultrasound image of normal appearing male genitalia
A 42-year-old G8P5116 female of Caucasian descent was evaluated at 22 weeks gestation due to a finding of gender discrepancy between karyotype and ultrasound.
Ultrasound image of normal appearing male genitalia
A 42-year-old G8P5116 female of Caucasian descent was evaluated at 22 weeks gestation due to a finding of gender discrepancy between karyotype and ultrasound.
Routine amniocentesis, performed at 16 weeks gestation for advanced maternal age, documented a 46, XX, normal female karyotype, while the pre-procedural ultrasound evaluation showed normal male genitalia.
Given this scenario, what diagnoses should be considered in the differential?
- Congenital adrenal hyperplasia (CAH)
- 46, XX testicular disorder of sex development (DSD)
- Maternal cell contamination (MCC)
- Androgen insensitivity (AI)
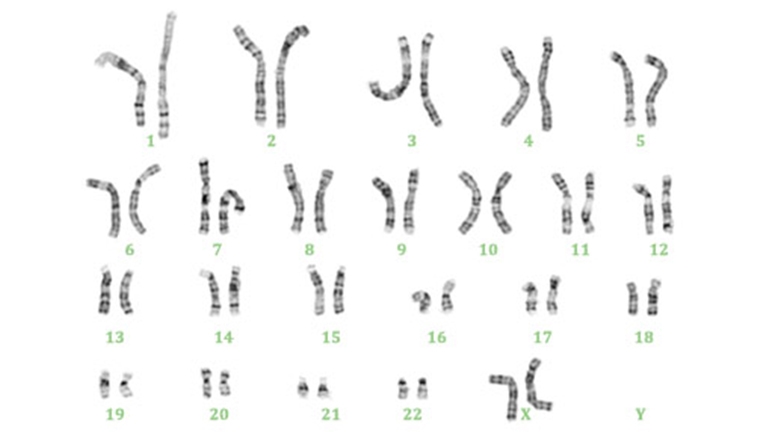 Image of a normal female karyotype. Image provided by the CHOP Cytogenomics Laboratory
Repeat amniocentesis, performed at 20 weeks gestation, confirmed the finding of a normal 46, XX karyotype and the presence of normal male external genitalia. Fluorescence in situ hybridization (FISH) studies demonstrated a copy of the SRY gene (sex-determining region of Y) on the short arm of one of the X chromosomes.
Image of a normal female karyotype. Image provided by the CHOP Cytogenomics Laboratory
Repeat amniocentesis, performed at 20 weeks gestation, confirmed the finding of a normal 46, XX karyotype and the presence of normal male external genitalia. Fluorescence in situ hybridization (FISH) studies demonstrated a copy of the SRY gene (sex-determining region of Y) on the short arm of one of the X chromosomes.
Evaluation
A detailed review of family, medical and pregnancy history was unremarkable for the presence of any known genetic conditions, structural anomalies and teratogenic exposures. Consanguinity was denied. Repeat targeted sonography revealed an appropriately grown fetus without apparent structural anomalies and normal-appearing male external genitalia.
Pregnancy and delivery course
The patient had a routine prenatal course and uneventful full-term vaginal delivery of a 3kg infant with normal external male genitalia. All newborn screening tests were within normal limits. A scrotal ultrasound revealed normally appearing bilaterally descended testicles. No Mullerian structures were identified on pelvic ultrasound. Infant and mother were discharged following a 48-hour hospital stay.
Questions
- What additional studies, if any, might be considered?
- How should this family be counseled?
- What are the long-term implications of this diagnosis for this child?
- Are there any other services that should become involved with this family?
Read on below for the answers.
Answers
Answers 1 and 2 and 3 might be considered in the differential, with 2 being the most likely. The most common form of congenital adrenal hyperplasia (CAH), 21-hydroxylase deficiency, typically presents with ambiguous genitalia on ultrasound and a female karyotype.
Individuals with 46, XX testicular disorder of sex development (46, XX testicular DSD), which is sometimes still referred to as 46, XX male syndrome, have a female karyotype, male external genitalia, two testicles, azoospermia and absence of Mullerian structures. The majority demonstrate the presence of SRY (sex-determining region of Y) within their genome, most commonly on one of the X chromosomes.
Maternal cell contamination (MCC) is also a possibility, although it commonly manifests as 46, XX/46, XY mosaicism.
The genotype-phenotype discordance in answer 4, androgen insensitivity (AI), is that of a normal male karyotype and normal female external genitalia.
- What additional studies, if any, might be considered? In view of rare reports of syndromic testicular DSD involving microdeletions of Xp on the derivative (der) X-chromosome as the consequence of the recombination event, microarray testing as well as FISH confirmatory testing was performed to rule out deletions within the steroid-sulphatase (STS) as well as Kallmann regions. Results indicated that no deletions were detected. In addition, the presence of SRY on Xp typically occurs de novo, but rare cases of inheritance have been described. For reassurance, both parents were karyotyped and had SRY FISH studies performed, which revealed results expected for a normal male and female.
- How should this family be counseled? The family should be counseled that the natural history of this presentation of 46, XX testicular DSD is expected to be that of a male phenotype and identity with concerns related to delayed puberty, hyphotrophic hypogonadism and likely infertility, but without learning and behavior problems or developmental delay.
- What are the long-term implications for this child? Long-term implications center on the above-mentioned issues of puberty and management of hyphotrophic hypogonadism. Testosterone replacement therapy might be indicated. Infertility is expected.
- Are there any other services that should become involved with this family? A multifaceted approach of clinical care and anticipatory guidance provided by a multidisciplinary team including urology, endocrinology and genetics, as well as psychology, is best suited to meet the needs of this population. Psychological support is important throughout, but especially so at and around the time of disclosure.
Reference
- Vilain, EJ. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. 46, XX Testicular Disorder of Sex Development. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014.

